Software, Algorithms, and Computational Methods
At the LSP, we pair innovative methods for data collection with cutting-edge computational methods and software. Our software methods enable a systems-level view of biology and pharmacology and cover various topics, such as the analysis of high-throughput drug screening data, biological knowledge assembly, and the analysis and visualization of multiplexed images.
Browse the tools below and visit the dedicated websites for each software to learn more. Generally, we release our code on GitHub under permissive open-source licenses (see Research Reproducibility).
Multiple-choice microscopy pipeline (MCMICRO)
MCMICRO is an open-source processing pipeline for multiplexed whole-tissue images and tissue microarrays. It consists of customizable modules that sequentially perform image processing and quantification steps, including stitching, registration, cell segmentation, single-cell quantification, and visualization. Each module is containerized with Docker, making it possible to deploy MCMICRO across various computing environments, including local machines, job-scheduling clusters, and cloud environments like AWS. MCMICRO is undergoing active development, and modules are regularly added and improved as part of the growing MCMICRO community.
Publication: Schapiro D, Sokolov A, Yapp C, Chen Y-A, Muhlich JL, Hess J, Creason AL, Nirmal AJ, Baker GJ, Nariya MK, Lin J-R, Maliga Z, Jacobson CA, Hodgman MW, Ruokonen J, Farhi SL, Abbondanza D, McKinley ET, Persson D, Betts C, Sivagnanam S, Regev A, Goecks J, Coffey RJ, Coussens LM, Santagata S, Sorger PK. MCMICRO: a scalable, modular image-processing pipeline for multiplexed tissue imaging. Nat Methods. 2022 Mar;19(3):311–315. DOI: 10.1038/s41592-021-01308-y. PMCID: PMC8916956
Minerva
Minerva is a suite of lightweight software tools that enables interactive viewing and fast sharing of large image data. With Minerva Author, users can import an image and generate a multi-waypoint, annotated Minerva Story that walks viewers through their data. Minerva Stories are hosted on the web and viewed through a web browser (no download required!), making them ideal for sharing large-scale image data as part of tissue atlases.
Publication: Rashid R, Chen, YA, Hoffer J, Muhlich JL, Lin JR, Krueger R, Pfister H, Mitchell R, Santagata S, Sorger PK. (2021). Narrative online guides for the interpretation of digital-pathology images and tissue-atlas data. Nature Biomedical Engineering, pp.1-12. DOI: 10.1038/s41551-021-00789-8. PMCID: PMC9079188
Hoffer J, Rashid R, Muhlich JL, Chen, YA, Russell D, Ruokonen J, Krueger R, Pfister H, Santagata S, Sorger PK. (2020). Minerva: a light-weight, narrative image browser for multiplexed tissue images. Journal of Open Source Software, 5(54), 2579. DOI: 10.21105/joss.02579. PMCID: PMC7989801
SCIMAP
SCIMAP is a scalable toolkit for analyzing spatial molecular data. SCIMAP is compatible with spatial datasets mapped to X-Y coordinates and can be integrated with many single-cell analysis toolkits. SCIMAP supports preprocessing, phenotyping, visualization, clustering, spatial analysis, and differential spatial testing, and can efficiently analyze large datasets containing millions of cells.
Publication: Nirmal AJ, Maliga Z, Vallius T, Quattrochi B, Chen AA, Jacobson CA, Pelletier RJ, Yapp C, Arias-Camison R, Chen YA, Lian CG, Murphy GF, Santagata S, Sorger PK. The spatial landscape of progression and immunoediting in primary melanoma at single-cell resolution. Cancer Discov. 2022 Jun 2;12(6):1518–1541. DOI: 10.1158/2159-8290.CD-21-1357. PMCID: PMC9167783
Nirmal AJ, Sorger PK. SCIMAP: A Python Toolkit for Integrated Spatial Analysis of Multiplexed Imaging Data. JOSS. 2024 May 29;9(97):6604.
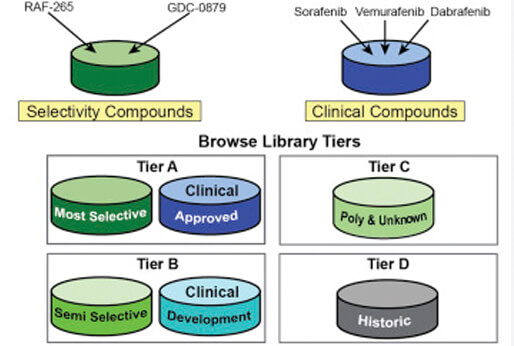
The Small Molecule Suite
The Small Molecule Suite (SMS) is a free, open-access tool. The goal of the SMS is to help scientists work with the targets of molecular probes, approved drugs, and other drug-like molecules, while understanding the complexity of polypharmacology — the phenomenon that acknowledges that virtually all molecules bind multiple proteins. The SMS combines data from the ChEMBL database with data from the Laboratory of Systems Pharmacology.
Publication: Moret N, Clark NA, Hafner M, Wang Y, Lounkine E, Medvedovic M, Wang J, Gray N, Jenkins J, Sorger PK. Cheminformatics Tools for Analyzing and Designing Optimized Small-Molecule Collections and Libraries. Cell Chem Biol. 2019 May 16;26(5):765-777.e3. DOI: 10.1016/j.chembiol.2019.02.018. PMCID: PMC6526536.
CyLinter
CyLinter is quality control software for multiplexed microscopy images that identifies and removes single cell data that has been corrupted by artifacts. Artifacts can result from optical aberrations (e.g., from contaminating lint or hair on the sample) or image-processing errors (e.g., errors in single cell segmentation) and can confound downstream analyses. CyLinter guides users through the process of removing noisy data in an interactive, visual environment and improves the quality of the resulting single cell data.
Publication: Baker GJ, Novikov E, Zhao Z, Vallius T, Davis JA, Lin JR, Muhlich JL, Mittendorf EA, Santagata S, Guerriero JL, Sorger PK. Quality control for single cell analysis of high-plex tissue profiles using CyLinter. bioRxiv; 2023. DOI: 10.1101/2023.11.01.565120 PMCID: PMC10634977.
Integrated Network and Dynamical Reasoning Assembler (INDRA)
INDRA (Integrated Network and Dynamical Reasoning Assembler) is an automated model assembly system. At the core of INDRA are its knowledge-level assembly procedures. INDRA draws on natural language processing systems and structured databases to assemble mechanistic knowledge into coherent models such as causal graphs and dynamical models. To do this, INDRA corrects systematic input errors, resolves redundancies, infers missing information, filters for scope, and assesses the reliability of the gathered knowledge.
Publication: Gyori BM, Bachman JA, Subramanian K, Muhlich JL, Galescu L, Sorger PK. From word models to executable models of signaling networks using automated assembly. Molecular Systems Biology. 2017 Nov 24;13(11):954. DOI: 10.15252/msb.20177651. PMCID: PMC5731347.
Bachman JA, Gyori BM, Sorger PK. Automated assembly of molecular mechanisms at scale from text mining and curated databases. Mol Syst Biol. 2023. May 9;19(5):e11325. DOI: 10.15252/msb.202211325. PMCID: PMC10167483.
ASHLAR (Alignment by Simultaneous Harmonization of Layer/Adjacency Registration)
ASHLAR (Alignment by Simultaneous Harmonization of Layer/Adjacency Registration) is an open-source Python tool that combines multi-tile microscopy images into high-dimensional mosaic images. For multi-cycle imaging methods (like CyCIF), ASHLAR also aligns images from different cycles with a high level of accuracy. ASHLAR can be used with virtually any unstitched microscope image file and multiplexed imaging method.
Publication: Muhlich JL, Chen YA, Yapp C, Russell D, Santagata S, Sorger PK. Stitching and registering highly multiplexed whole slide images of tissues and tumors using ASHLAR. Bioinformatics. 2022 Aug 16;btac544. DOI: 10.1093/bioinformatics/btac544. PMCID: PMC9525007
ShinyDepMap
shinyDepMap is a web-tool to explore the Cancer Dependency Map (DepMap) data of the Broad Institute (version 19q3). We combined the DepMap CRISPR and RNAi dependency data into a unified score, and built this easy-to-use tool. shinyDepMap can be used to predict the efficacy and selectivity of future drugs with a known target gene, identify targets of highly selective drugs, identify maximally sensitive cell lines for testing a drug, identify similar targets that may be easier to drug (i.e., “target hop”), and identify pathways necessary for cancer cell growth and survival.
Publication: Shimada K, Bachman JA, Muhlich JL, Mitchison TJ. shinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. Elife. 2021 Feb 8;10. DOI: 10.7554/eLife.57116. PMCID: PMC7924953.
Systemic Lymphoid Architecture Response Assessment (SYLARAS)
SYLARAS (SYstemic Lymphoid Architecture Response ASsessment) is a platform for interrogating the systemic immune response to a disease or therapy. SYLARAS encompasses an experimental method for collecting multiplexed flow cytometry data, computer-assisted pre-processing steps that deconvolve and filter data, and a computational analysis regime that generates and displays longitudinal immunophenotyping data on a per-cell type basis. The ability of SYLARAS to generate and display interpretable systemic immune response data makes it a broadly useful preclinical research tool.
Publication: Baker GJ, Muhlich JL, Palaniappan SK, Moore JK, Davis SH, Santagata S, Sorger PK. SYLARAS: A Platform for the Statistical Analysis and Visual Display of Systemic Immunoprofiling Data and Its Application to Glioblastoma. Cell Syst. 2020 Sep 23;11(3):272-285.e9. DOI: 10.1016/j.cels.2020.08.001. PMCID: PMC7565356.