LSP Data and Methods
LSP investigators use highly multiplexed assays that collect many pieces of data from cell extracts, cultured cells, and whole tissues. Our datasets often include proteomics, live and fixed-cell imaging, transcript profiling, and cytokine profiling. We also develop many new methods for data collection and analysis.
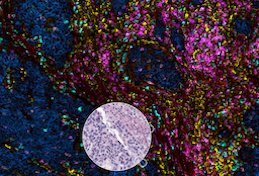
Harvard Tissue Atlas
The Harvard Tissue Atlas (HTA) assembles image and -omic datasets into precise 3D molecular maps of cell types, states, and interactions between cells and acellular structures. Our atlases bring together multiple research projects examining normal and diseased tissue from human and animal models, with an emphasis on cancer. We aim to better understand disease initiation and development to enable the next generation of diagnostic molecular tests and precision medicine approaches.
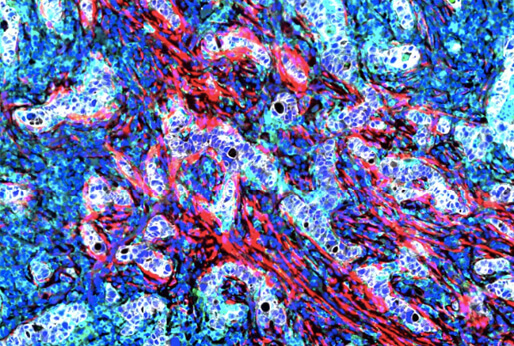
Cyclic Immunofluorescence (CyCIF)
CyCIF (cyclic immunofluorescence) is a robust and inexpensive method for highly multiplexed immunofluorescence imaging using standard instruments and reagents. Tissue-based (t-) CyCIF generates multiplexed images of fixed, paraffin-embedded (FFPE) samples using an iterative process in which conventional low-plex fluorescence images are repeatedly collected from the same sample and then assembled into a high dimensional representation. Highly multiplexed images of intact tumor architecture can be used to quantify signal transduction cascades, measure the levels of tumor antigens, determine precise immune phenotypes, and more.
Publication: Lin J-R, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, Sorger PK. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife. 2018 Jul 11;7. PMCID: PMC6075866.
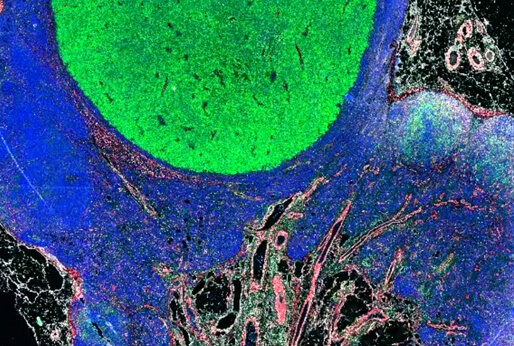
Orion
Orion is a method for collecting one-shot 18-plex immunofluorescence images and diagnostic-grade H&E images from the same samples. The Orion method was developed in collaboration with RareCyte Inc. and uses a specialized microscope and fluorescent antibodies (known as ArgoFluors™), which can be imaged simultaneously and spectrally unmixed. We show that same-slide H&E and IF images provide complementary information that can be used to train ML models that effectively predict cancer progression.
Publication: Lin JR, Chen YA, Campton D, Cooper J, Coy S, Yapp C, Tefft JB, McCarty E, Ligon KL, Rodig SJ, Reese S, George T, Santagata S, Sorger PK. High-plex immunofluorescence imaging and traditional histology of the same tissue section for discovering image-based biomarkers. Nat Cancer. 2023 Jul;4(7):1036–1052. DOI: 10.1038/s43018-023-00576-1. PMCID: PMC10368530.
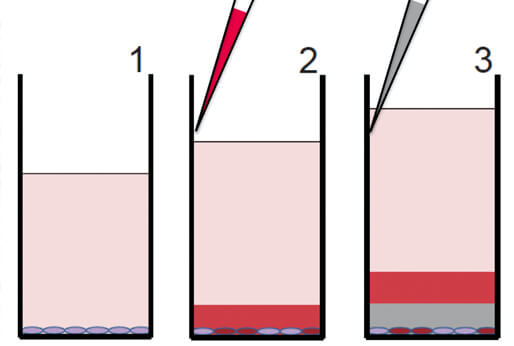
DeepDyeDrop
The Dye Drop assay is a versatile, low-cost, multiplexed microscopy method for obtaining detailed single-cell viability and cell cycle data. The Dye Drop method uses a set of incrementally more dense solutions to prevent cell loss and improve the consistency of cell perturbation experiments. Dye Drop can also be combined with CyCIF to yield further molecular and spatial information. The method is paired with computational analysis tools that calculate cell state and growth rate metrics from the high throughput data.
Publication: Mills CE, Subramanian K, Hafner M, Niepel M, Gerosa L, Chung M, Victor C, Gaudio B, Yapp C, Nirmal AJ, Clark N, Sorger PK. Multiplexed and reproducible high content screening of live and fixed cells using Dye Drop. Nature Communications. 2022 Nov 14;13(1):6918. DOI: 10.1038/s41467-022-34536-7. PMCID: PMC9663587.
HMS LINCS Database
The HMS LINCS Database is a public resource for browsing, searching, and downloading all fully annotated datasets generated through the HMS LINCS Center along with experimental and analytical methods. The database contains >350 datasets from imaging, transcriptomic, and proteomic experiments as well as in vitro small molecule affinity assays.
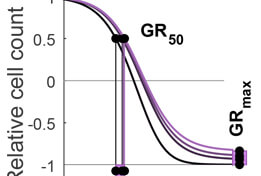
GR Calculator/Browser
The Growth Rate inhibition (GR) Calculator (grcalculator.org) is an open source set of Python, R and online tools for quantifying the responses of cancer cells to drugs in a manner that corrects for the confounding effects of variable cell proliferation rates. Response metrics computed from GR data include GR50 and GRmax and are direct analogs of familiar IC50 and Emax response measures.
Publication: Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods. 2016 Jun 1;13(6):521–527. DOI: 10.1038/nmeth.3853. PMCID: PMC4887336.
Hafner M, Niepel M, Sorger PK. Alternative drug sensitivity metrics improve preclinical cancer pharmacogenomics. Nat Biotechnol. 2017 Jun 7;35(6):500–502. DOI: 10.1038/nbt.3882. PMCID: PMC5668135.
ProteinNet
ProteinNet is a standardized data set for machine learning of protein structure. It provides protein sequences, structures (secondary and tertiary), multiple sequence alignments (MSAs), position-specific scoring matrices (PSSMs), and standardized training / validation / test splits. ProteinNet builds on the biennial CASP assessments, which carry out blind predictions of recently solved but publicly unavailable protein structures, to provide test sets that push the frontiers of computational methodology. It is organized as a series of data sets, spanning CASP 7 through 12 (covering a ten-year period), to provide a range of data set sizes that enable the assessment of new methods in relatively data-poor and data-rich regimes.
Publication: AlQuraishi M. ProteinNet: a standardized data set for machine learning of protein structure. BMC Bioinformatics. 2019 Jun 11;20(1):311. DOI: 10.1186/s12859-019-2932-0. PMCID: PMC6560865.

DRIAD
DRIAD (Drug Repurposing In Alzheimer’s Disease) is a machine learning framework for identifying potential associations between Alzheimer’s Disease pathology and individual genes. DRIAD was applied to data from perturbation experiments performed in the LSP using differentiated human neural cell cultures and 80 FDA-approved and clinically tested drugs, to nominate existing drugs that could be potentially be repurposed for Alzheimer’s treatment.
Publication: Publications: Rodriguez S, Hug C, Todorov P, Moret N, Boswell SA, Evans K, Zhou G, Johnson NT, Hyman BT, Sorger PK, Albers MW, Sokolov A. Machine learning identifies candidates for drug repurposing in Alzheimer’s disease. Nat Commun. 2021 Feb 15;12(1):1033. DOI: 10.1038/s41467-021-21330-0. PMCID: PMC7884393.
CancerTrials.io
CancerTrials.io contains imputed individual participant data (IPD) from previously published oncology research clinical trials (RCTs). The site also contains various types of analysis performed on these data.
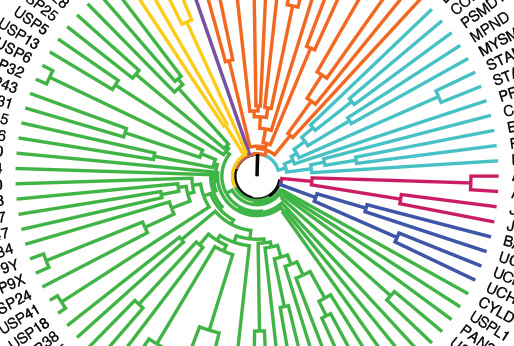
DUB Portal
DUB (Deubiquitinating enzymes) portal provides the integration and analysis of publicly available resources as well as newly collected transcriptomic data following DUB knockout or inhibition to facilitate the exploration of DUB function in oncology.
Publication: Doherty LM, Mills CE, Boswell SA, Liu X, Hoyt CT, Gyori B, Buhrlage SJ, Sorger PK. Integrating multi-omics data reveals function and therapeutic potential of deubiquitinating enzymes. Elife. 2022 Jun 23;11:e72879. DOI: 10.7554/eLife.72879. PMCID: PMC9225015